目的
- AutoDock Vinaを用いたドッキング計算方法の流れを理解する
AutoDock Vinaを選定した理由
- ドッキングソフトウェアはDOCK, AutoDock, AutoDock Vinaなど様々あるがそれぞれ一長一短である。これまでにDOCKは使ったことがあるが, 系に応じて適切な手法を選択できるように別のソフトウェアも使ってみたいと考えていた。
- AutoDock Vinaはempiricalかつknowledge-basedなポテンシャルを採用しているため高速かつ精度の良い計算が可能である。また, 業界スタンダードであるためドキュメントが豊富な上に使用が簡単である(らしい)。そこで今回はAutoDock Vinaで一通りの計算を実行してみる。
参考
必要なツールのセットアップ
必要なツールは本来Pymol, Autodock Vina, Autodock MGL toolsの3つ…なのだがAutodock MGL toolsのGUIはMac OS Catalinaで動かない(PythonスクリプトはPython2対応で書かれているもののきちんと動く)
追記:Dockerを導入することでMacOS上でLinux版を動かすことができた。詳しくはmac-osx-catalinaに導入したdocker上でautodock-toolsを動かすを参照。
そこで今回はChimeraを補完的に用いることにした。Pymol, MGL toolsおよびChimeraを手元のMacBook Proへ, AutoDock VinaはLinuxのスパコンへインストールした。
Chimera
Chimera本体をインストーラからインストールする。簡単。
AutoDock Vina
最新版をダウンロードして解凍する。コンパイル済みなので解凍するだけで動く。
Pymol
元々Macに入っていたので割愛。
AutoDock MGL tools
最新版を取ってきて解凍する。GUI部分はMac OSX Catalinaで動かないが使わないので問題ない。
計算の実施
計算の流れは大まかに以下のようになる。必要なツールを括弧内に記載いた。
- 計算対象のリガンドおよびタンパク質をPDB形式で書き出す (Pymol)
- 計算対象のタンパク質とリガンドに前処理を実施 (MGL tools)
- .confファイルを作成し計算オプションを指定 (Chimera, テキストエディタ)
- ドッキングを実施 (AutoDock Vina)
(Pymol) 計算対象のリガンドおよびタンパク質をPDB形式で書き出す
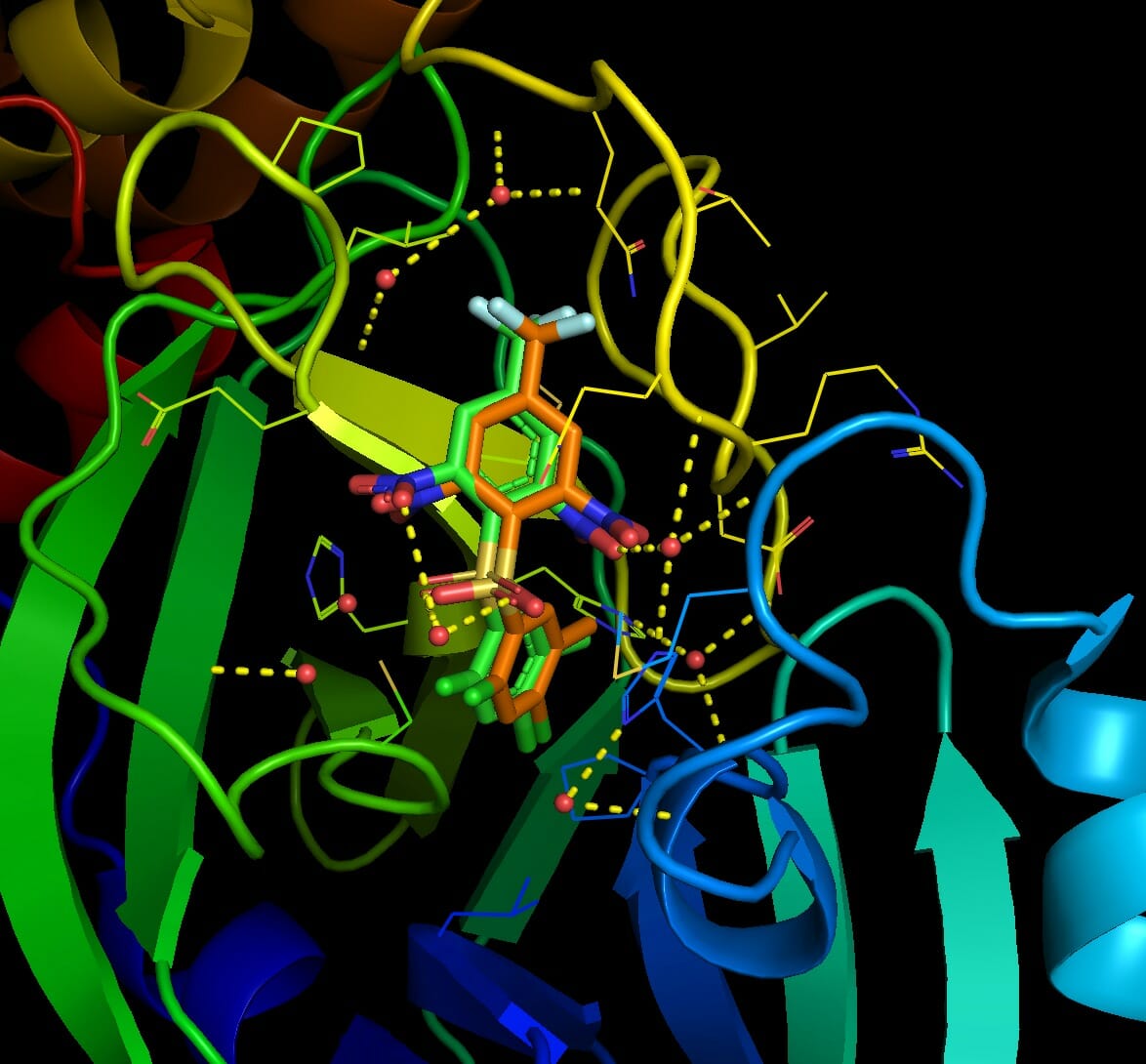
ドッキング計算では計算結果と結晶構造のリガンドを重ね合わせることで計算手法の妥当性を検討することがとても重要である。
今回はリガンドの結晶構造が既知のSARS-Cov-1のMain protease(PDB ID: 2GZ7)をターゲットタンパク質とし, 計算結果と結晶構造との比較を行う。このタンパク質は2020年現在を世界を大きく騒がせているSARS-Cov-2(通称COVID-19)のMain proteaseと相同性が非常に高く興味深い対象である。
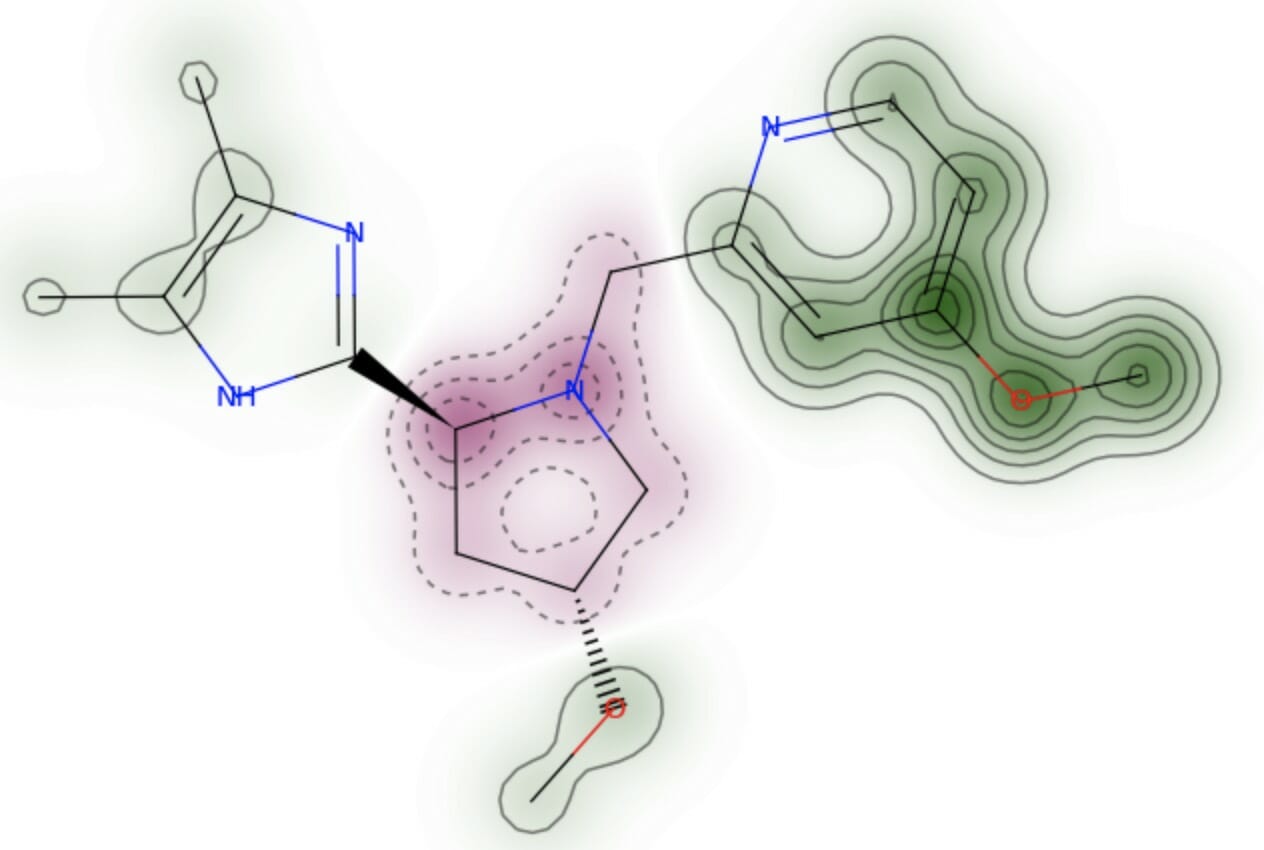
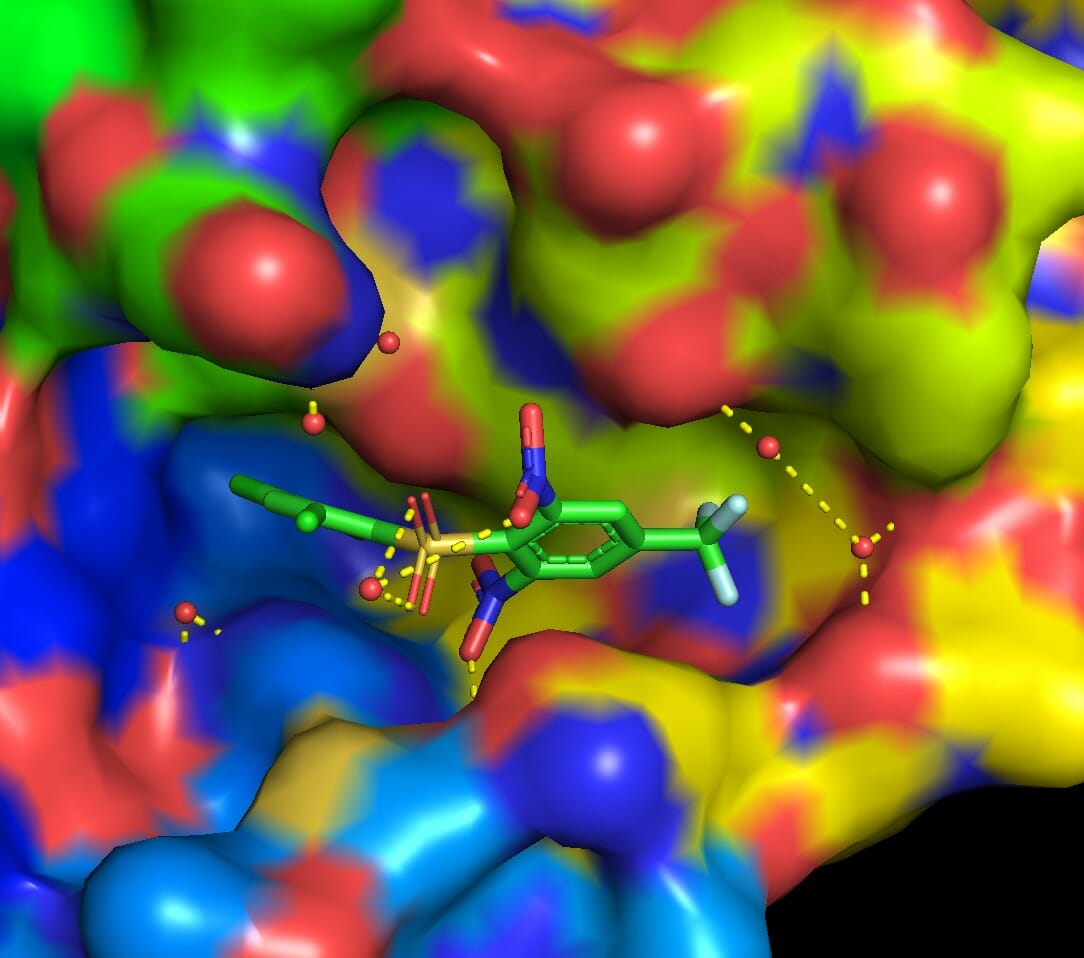
以下に結合サイトをPymolで図示した。リガンドが結合サイト内のアミノ酸残基および水分子と水素結合を形成している。このことは水分子がリガンドの結合に重要な役割を果たしている可能性を示唆するが, 今回は計算を簡略化するために水分子は全て取り除いた。
PyMOL>remove solvent上のコマンドで水を取り除き, タンパク質およびリガンドをそれぞれligand.pdbおよびprotein.pdbというファイル名で保存した。詳しいPymolの操作方法は別のサイトを御覧いただきたい。
(MGL Tools) 計算対象のタンパク質とリガンドに前処理を実施
前述のリガンドおよびレセプターのPDBファイルをMGL Toolsで処理する。MGL ToolsはPython 2で書かれているためPython3だと動かないので注意。自分はanacondaでpython2.7の環境を作成してその中で動かした。
まず標的リガンドであるligand.pdbを以下のコマンドで処理する。”-A bonds_hydrogen”オプションを追加することでMGL toolsが自動的にリガンドの結合情報を変更し水素原子を付加してくれる。処理後はligand.pdbqtというファイルが生成され, これをAutodock Vinaのインプットとして用いる。
/(Path_to_MGLtools)/mgltools_i86Darwin9_1.5.6/MGLToolsPckgs/AutoDockTools/Utilities24/prepare_ligand4.py \
-l ligand.pdb -A bonds_hydrogen 次に標的タンパク質であるprotein.pdbも同様に処理する。得られたprotein.pdbqtをAutodock Vinaのインプットとして用いる。
/(Path_to_MGLtools)/mgltools_i86Darwin9_1.5.6/MGLToolsPckgs/AutoDockTools/Utilities24/prepare_receptor4.py \
-r protein.pdb -A bonds_hydrogen AutoDock Vinaは計算時に電荷情報を自動的に追加するため, インプットファイルで電荷情報を追加する必要はない(“Why don’t the results change when I change the partial charges?”参照)。
また, AutoDock VinaはUnited Atom modelを採用しているため, Non-polar hydrogenはAutoDock Vinaの計算実行時に全て削除される。一方でAutoDock Vinaは自動的に水素原子を付加してくれるわけではないので, polar hydrogenの位置を把握するためにも入力ファイルには水素原子の結合情報を与える必要がある。
(Chimera, テキストエディタ) .confファイルを作成し計算オプションを指定
AutoDock Vinaでは計算時にポケットの位置を指定する必要がある。ポケットの位置は.confファイル内に記入する。
まずChimeraでligand.pdbおよびprotein.pdbを開く。MGL Toolsで作成したpdbqtファイルは読み込めないので注意。次にTools -> AutoDock Vinaからポケットの位置を指定する。ligand.pdbファイル内の原子の座標を参考に”Receptor search volume options”からボックスの大きさおよび中心を指定し, ポケットが十分覆われるようにする。
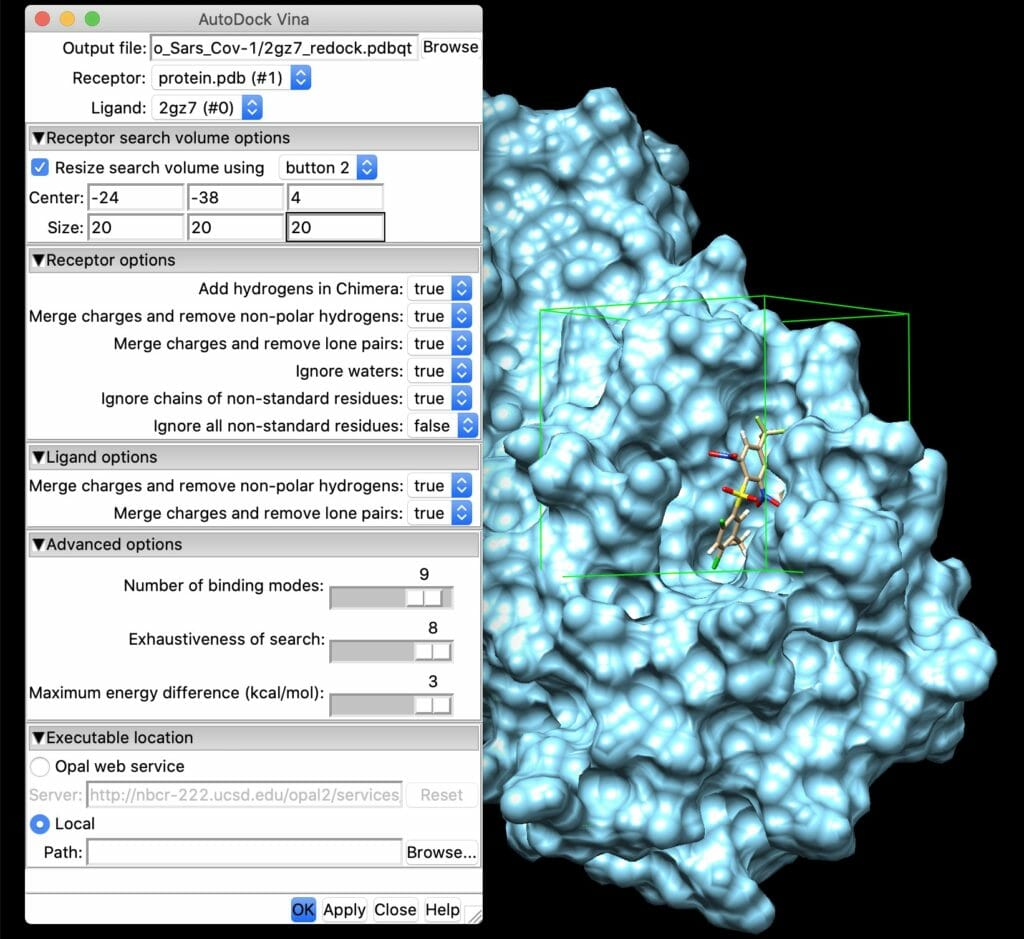
前述のボックスの座標情報を元にAutoDock Vinaの計算に必要な.confファイルをテキストエディタで作成する。以下に作成したautodock.confの例を示す。
center_x = -28.00
center_y = 38.00
center_z = 4.00
size_x = 20.00
size_y = 20.00
size_z = 20.00
energy_range = 3
exhaustiveness = 8
num_modes = 9その他のオプションについて説明する。いずれの値もAutoDock Vinaのデフォルト値に設定した。
energy_rangeはベストスコアから何kcal/molまでの結果を出力するかを意味する。具体的にはenergy_range = 3ではベストスコアが-9.0 kcal/molの場合, -6.0 kcal/molまでの結果が出力される。
exhaustivenessはエネルギー地形をどれだけ徹底的に探索するかを示している(詳細)。大きくするほど計算時間がかかるがグローバルミニマムを逃す確率が下がる。
num_modesは最大の出力結果数を示す。9なら9つのモードが出力される。
(AutoDock Vina) ドッキングを実施
ついにドッキングを実施する。先ほど作成した2つの.pdbqtファイルおよびconfファイルをLinuxに転送。以下のコマンドでシミュレーションを実施する。
/(path_to_vina)/vina --receptor receptor.pdbqt --ligand ligand.pdbqt \
--config autodock.conf --out autodock.pdbqt --log autodock.log計算はAMD Ryzen7 3800Xの32並列で約10秒で終了。はやい。以下のRMSDはベストスコアのもの(1番目のmode)に対する相対値として計算されるもので, 結晶構造に対するRMSDではないので注意。
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -7.1 0.000 0.000
2 -7.0 1.554 2.642
3 -7.0 4.893 6.867
4 -6.9 4.420 6.587
5 -6.9 1.526 3.694
6 -6.8 3.932 5.225
7 -6.8 4.876 7.154
8 -6.7 4.940 6.864
9 -6.7 5.309 7.497Code language: JavaScript (javascript)結果のautodock.pdbqtはPymolで可視化できる。
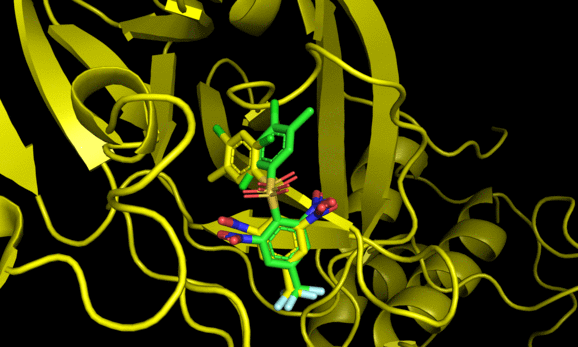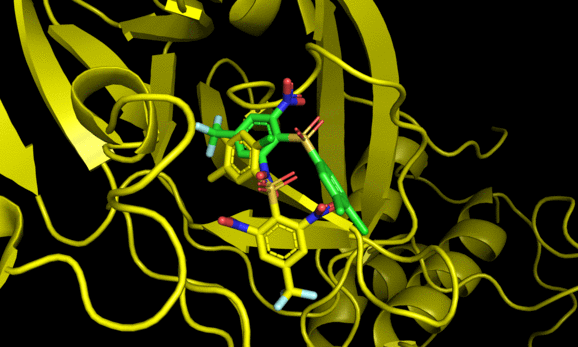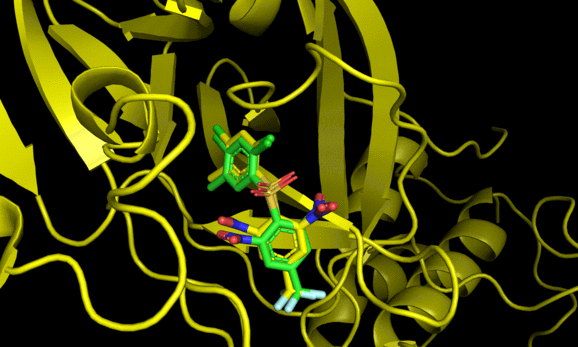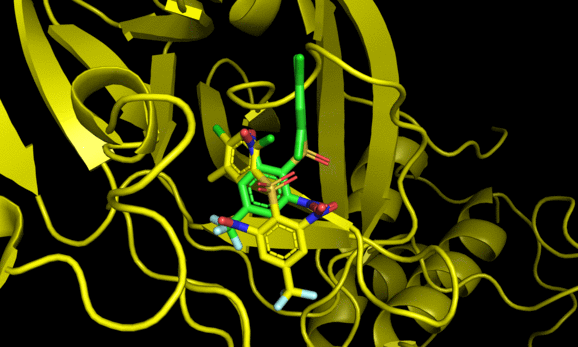
2番目と6番目のposeが結晶構造をよく再現した一方でそれ以外の構造は差異が大きい。6番目の結果にも関わらず結晶構造との再現度が高い辺り, スコアリング順が良い = ドッキング構造が良い, では必ずしもないことがわかる(参考)。
とはいえ正解のドッキングポーズが結果にきちんと含まれていることから, 今回の系に関してはin sillicoのアプローチが有効である可能性がある。今回の標的タンパク質と相同性が高いSARS-Cov-2のMpro阻害薬探索においてもドッキング計算は一定の有効性がありそうだ。